JAK Inhibitors in MyelofibrosisPatients without the JAK2 V617F mutation benefit equally from JI therapy as those with the mutation (11 faculty)
         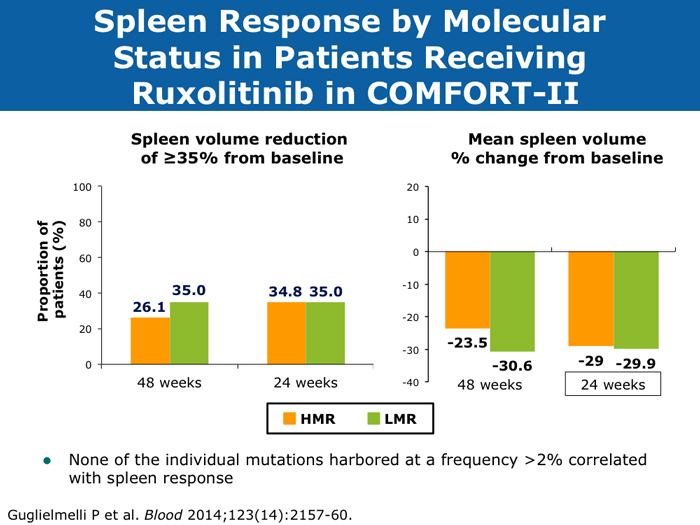 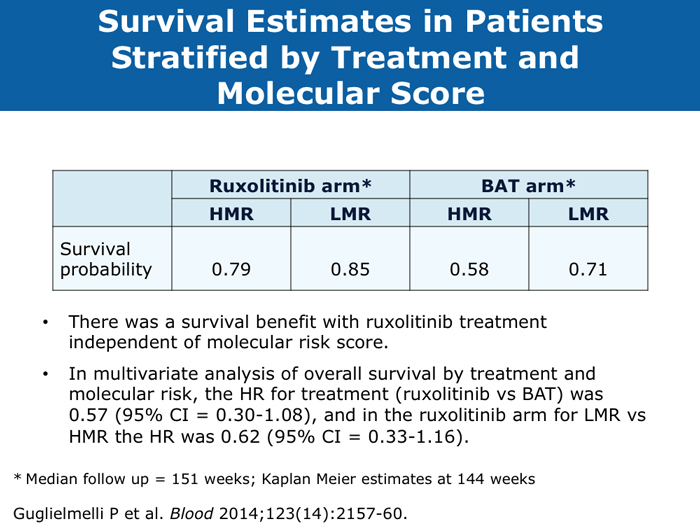 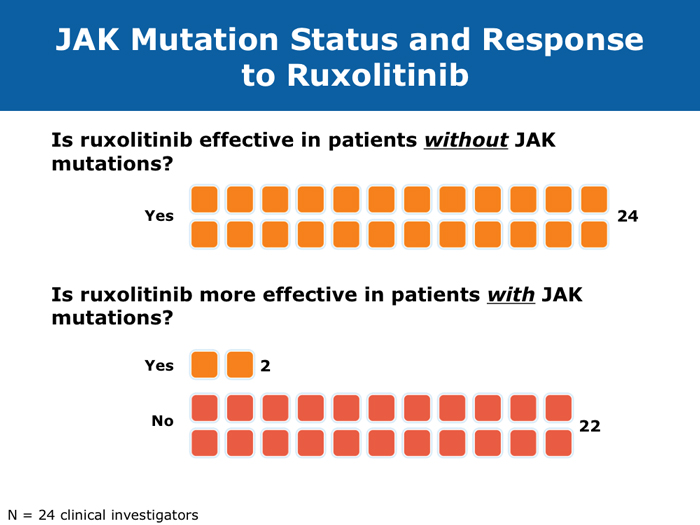 Dueck AC et al. Cytokine profile changes in 309 myelofibrosis patients: Comparison of JAK1/JAK2 inhibitor therapy vs placebo — Correlative analysis from the Comfort-I trial. Proc ASH 2013;Abstract 4074. Guglielmelli P et al; COMFORT-II Investigators; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative (AGIMM) Investigators. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood 2014;123(14):2157-60. Abstract Guglielmelli P et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 2011;118(19):5227-34. Abstract Klampfl T et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369(25):2379-90. Abstract Kralovics R et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005;352(17):1779-90. Abstract Levine RL. Another piece of the myeloproliferative neoplasms puzzle. N Engl J Med 2013;369(25):2451-2. Abstract Nangalia J et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369(25):2391-405. Abstract Squires M et al. The relationship between cytokine levels and symptoms in patients (Pts) with myelofibrosis (MF) from COMFORT-II, a phase 3 study of ruxolitinib (RUX) vs best available therapy (BAT). Proc ASH 2013;Abstract 4070. Initial evaluation of a patient with MF should include cytogenetics if obtainable and formal assessment of international prognostic score (11 faculty)
Jorge E Cortes, MD DR CORTES: I do believe myelofibrosis is underdiagnosed. You may have a patient who has some cytopenias only, typically an older patient, and that patient may or may not have an enlarged spleen. If the disease is advanced the spleen will be enlarged, but sometimes in the early stages it is not. It may be trickier to establish whether this is primary myelofibrosis or post-polycythemia vera or post-essential thrombocythemia myelofibrosis. I believe the diagnosis is more difficult in the earlier stages of myelofibrosis when it’s probably Grade I or perhaps Grade II, when some dysplastic features appear in the bone marrow. The disease can resemble myelodysplastic syndromes. If you are seeing a patient in the clinic today for the first time who already has myelofibrosis, you may not know if the patient’s history includes essential thrombocythemia or polycythemia vera. Sometimes examining the bone marrow may help you — that may reveal some features that suggest that it’s evolving from a precursor, but many times you simply won’t be able to make that diagnosis. In practical terms it probably doesn’t make much of a difference because nowadays you would probably treat in a similar way. The International Prognostic Scoring System (IPSS) and the Dynamic International Prognostic Scoring System (DIPSS) are useful tools. Like all prognostic classifications for any disease, they’re not perfect, but I believe they’re useful because they can clearly identify patients who are more likely to have good long-term outcomes and patients who are not expected to have good long-term outcomes, and they can guide your treatment decisions in an important way. For example, if you had a patient with myelofibrosis who was young and was a potential candidate for stem cell transplant but was at low risk by the DIPSS, then you definitely wouldn’t want to consider transplant because the expected survival is good. If the patient is considered to be at high risk, then you start considering transplant right away. Myelofibrosis is not a condition for which transplant is a straightforward consideration. These classifications can help you with these definitions. Alessandro M Vannucchi, MD DR VANNUCCHI: I believe myelofibrosis may be misdiagnosed quite often. Making the diagnosis of true myelofibrosis versus fibrosis in the bone marrow can be difficult. For this reason advanced-phase polycythemia vera or essential thrombocythemia can be mistakenly diagnosed as myelofibrosis, and the reverse is also true — patients with myelofibrosis can be diagnosed as having either of these 2 diseases. Also, I have seen older patients with anemia of unknown origin receive an incorrect diagnosis of myelofibrosis. In the diagnosis of myelofibrosis, obtaining a bone marrow biopsy is mandatory but the oncologist needs to obtain additional clinical information — for example, palpability of the spleen and patient age. In diagnosing myelofibrosis, obtaining a bone marrow biopsy is essential. Cytogenetic testing should also be routinely performed because the results can suggest treatment strategies for younger patients with unfavorable cytogenetics and transplant options. Cytogenetic testing is not always feasible because of dry taps of the bone marrow, and it is not easily performed on peripheral blood. We use FISH analysis for most of the common abnormalities. I also order analyses of LDH levels and MPL gene mutations. I order laboratory tests for erythropoietin levels only when a patient for whom I am considering treatment with recombinant human erythropoietin has anemia.  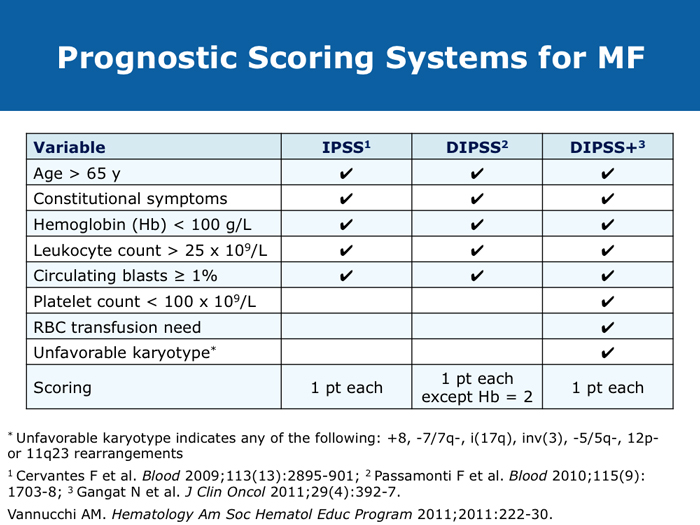 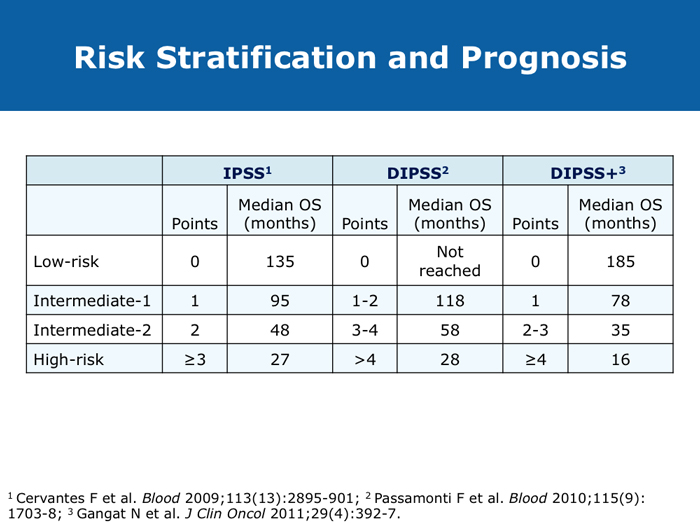 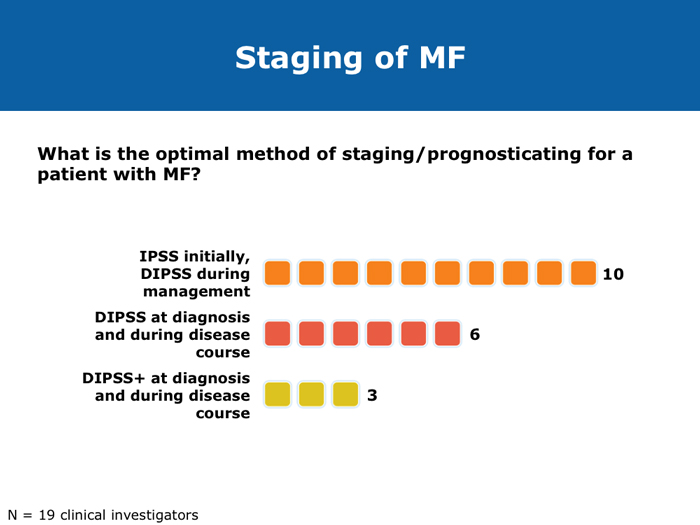 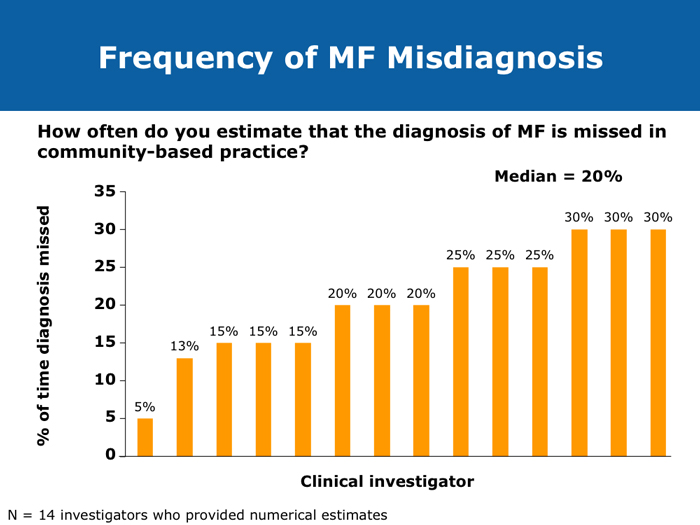 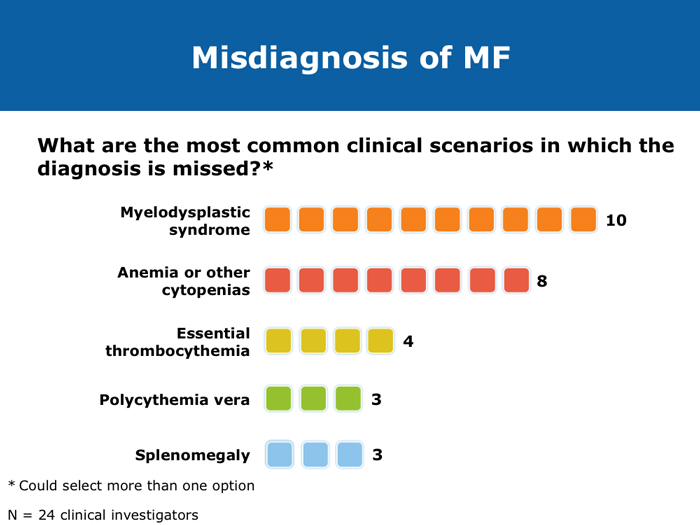  Cervantes F et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009;113(13):2895-901. Abstract Gangat N et l. DIPSS plus: A refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol 2011;29(4):392-7. Abstract Hussein K et al. International Prognostic Scoring System-independent cytogenetic risk categorization in primary myelofibrosis. Blood 2010;115(3):496-9. Abstract Kvasnicka HM. WHO classification of myeloproliferative neoplasms (MPN): A critical update. Curr Hematol Malig Rep 2013;8(4):333-41. Abstract Passamonti F et al. Impact of ruxolitinib on the natural history of primary myelofibrosis: A comparison of the DIPSS and the COMFORT-2 cohorts. Blood 2014;123(12):1833-5. Abstract Passamonti F et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010;115(9):1703-8. Abstract Passamonti F et al. Dynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosis. Blood 2010;116(15):2857-8. Abstract Yi CA et al. Discrepancy in diagnosis of primary myelofibrosis between referral and tertiary care centers. Leuk Res 2014;38(1):91-4. Abstract JIs do more than palliate symptoms (10 faculty)
John O Mascarenhas, MD DR MASCARENHAS: The availability of ruxolitinib has changed the treatment landscape. Ruxolitinib is effective in palliating symptoms and reducing splenomegaly, and some evidence indicates that prolonged therapy for 24 to 48 months may lead to retardation of fibrosis in the marrow. That’s an interesting finding with compelling implications. Despite the fact that the COMFORT-I and COMFORT-II trials were for intermediate- and high-risk myelofibrosis, I believe that patients with symptomatic low-risk disease can benefit from ruxolitinib. For patients with platelet counts lower than 50,000/µL, ruxolitinib is not an option. DR LOVE: In your clinical experience, what’s the minimal dose to achieve significant symptom palliation? DR MASCARENHAS: From the trials and from my own experience, you get the most “bang for your buck” in terms of symptom improvement at a dose of 10 mg BID. I don’t typically notice a tremendous improvement if the dose is increased to 15 mg BID. The caveat, however, is that for some people the itching can be better controlled at higher doses. I have 1 patient who’s receiving 25 mg BID, which is unusual. She’s maintained her platelet count for several years on this dose. When we reduce the dose her itching and bone pain start to come back. But for the majority of patients in terms of fatigue, anorexia and many of the symptoms, at 10 mg BID you’re achieving improvement. I don’t always find that a dose increase to 15 mg BID gets you that much further except in terms of achieving a deeper spleen reduction. Some data suggest that the deeper the spleen reduction, the more likely the patient is to derive a survival benefit. I believe those data are a little premature if you’re trying to reduce the spleen size. But for symptom improvement, 10 mg BID usually is a good dose. Elias Jabbour, MD DR JABBOUR: A question that I’m frequently asked is, “Should I start all patients with myelofibrosis on front-line treatment with ruxolitinib in light of its effect on survival?” The debate centers on the fact that most of the patients in the COMFORT studies had high-risk disease. My answer is that I use it for every patient who is symptomatic. The FDA label indications for ruxolitinib are the IPSS intermediate-1 disease or higher, and all patients with symptoms have at least intermediate-1-risk myelofibrosis. DR LOVE: Do any patients with lower-risk myelofibrosis have symptoms? DR JABBOUR: Yes. They can have inflammation or bone aches — not necessarily an enlarged spleen. David P Steensma, MD DR STEENSMA: The indications for treatment with ruxolitinib include a symptomatic patient, a patient whose spleen is becoming problematic and a patient with cytopenias of concern — patients who are nearing the level at which red blood cell transfusion is necessary, for whom the neutrophil count is dropping and platelet transfusion is required or bleeding complications occur. Patients who have low-risk disease and are minimally symptomatic can simply be observed and perhaps take a baby aspirin because a thrombotic risk exists with myelofibrosis. For those patients, the natural history is measured in many years. It’s not clear that we do them any good with treatment. Jerry L Spivak, MD DR SPIVAK: If you evaluate the natural history of myelofibrosis, you see that asymptomatic patients fare well, and I like to save my ammunition for when they are not faring well, so I would not administer a drug that could have toxicity but will not make them feel better. I wait to start treatment with ruxolitinib until patients are symptomatic.  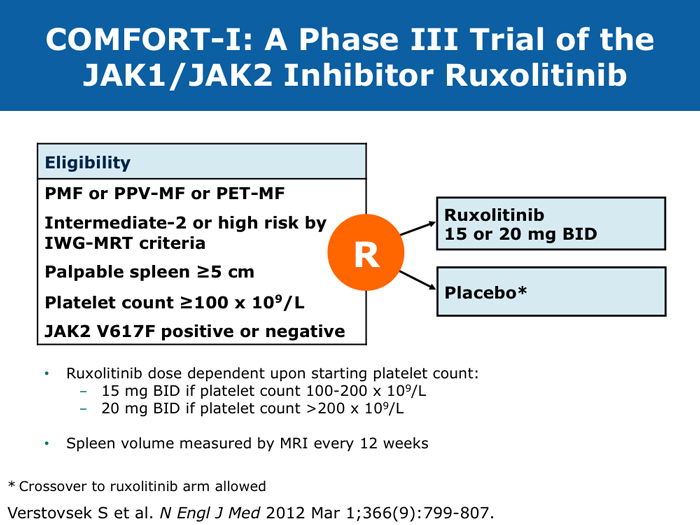 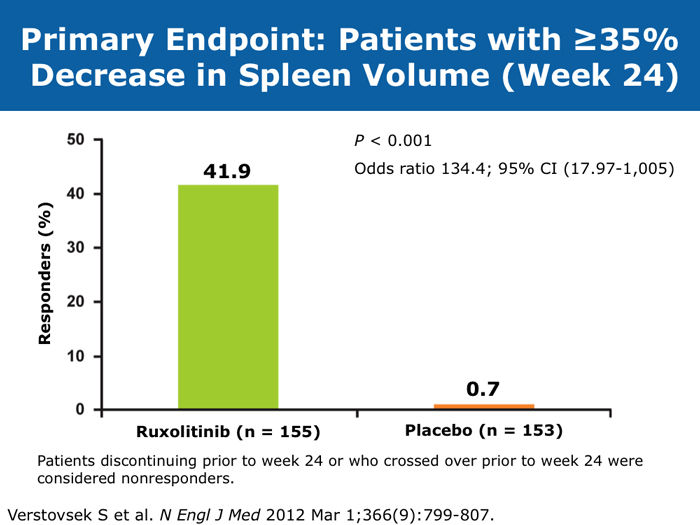 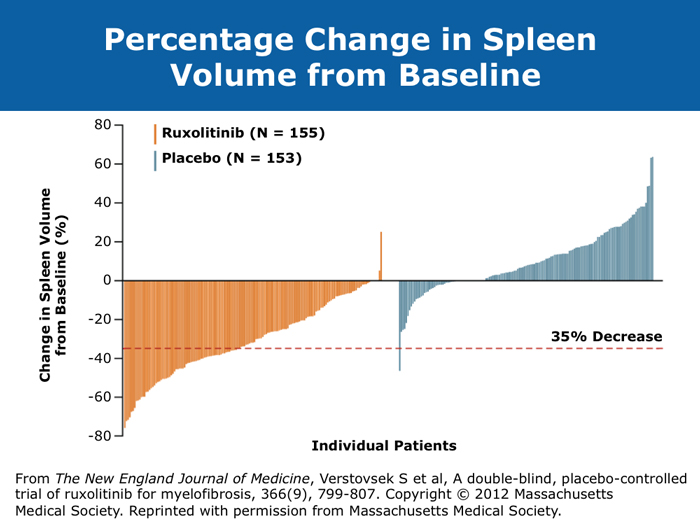 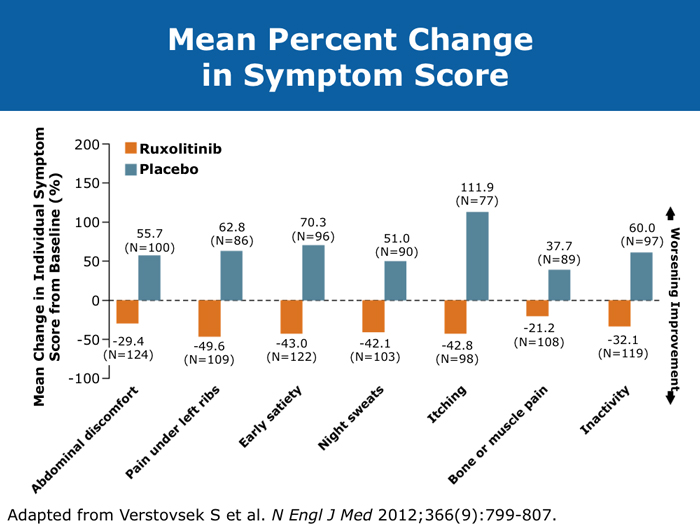 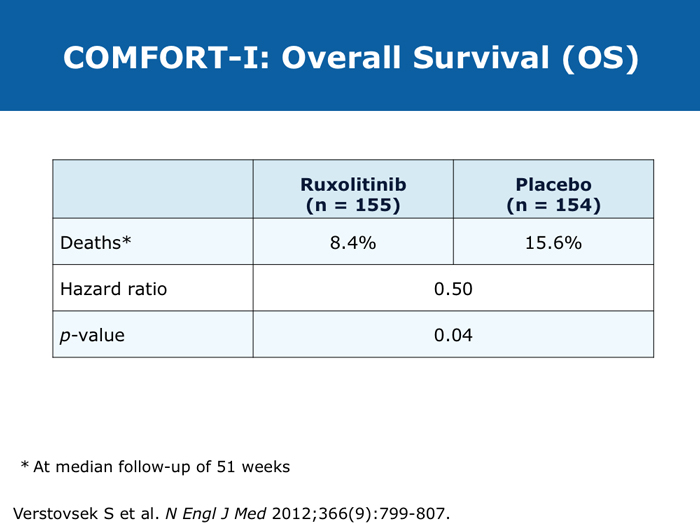 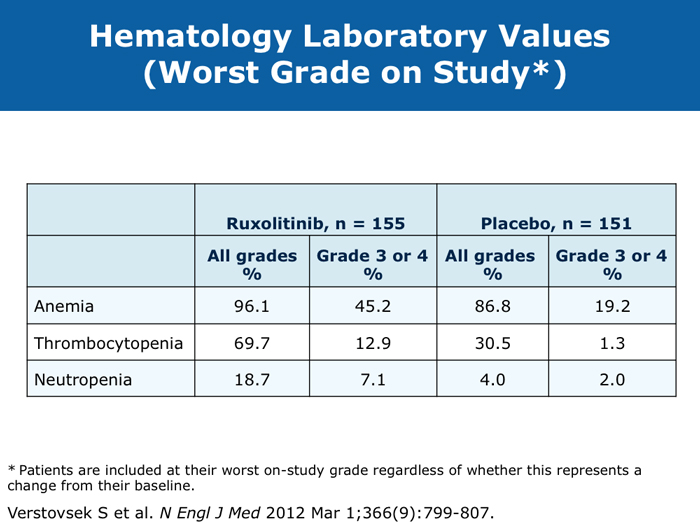  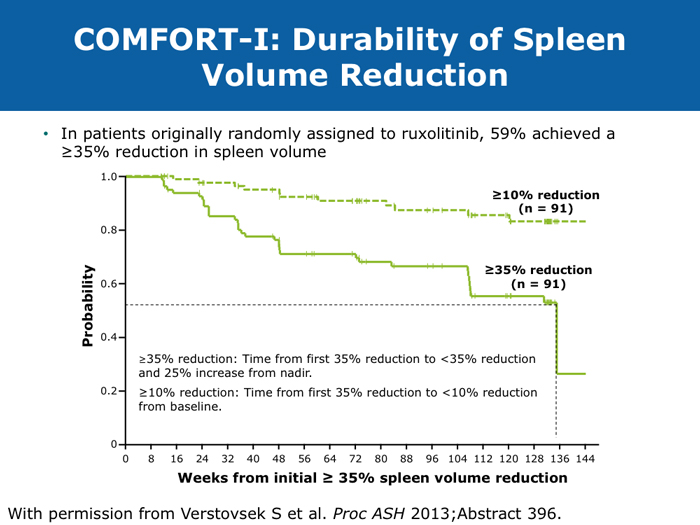 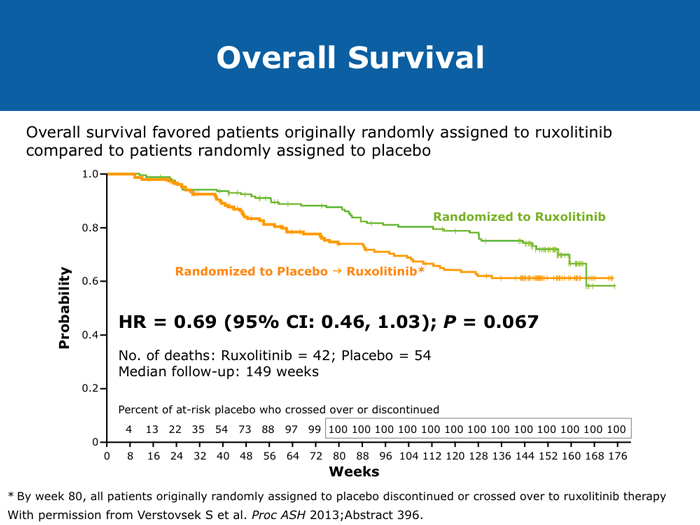 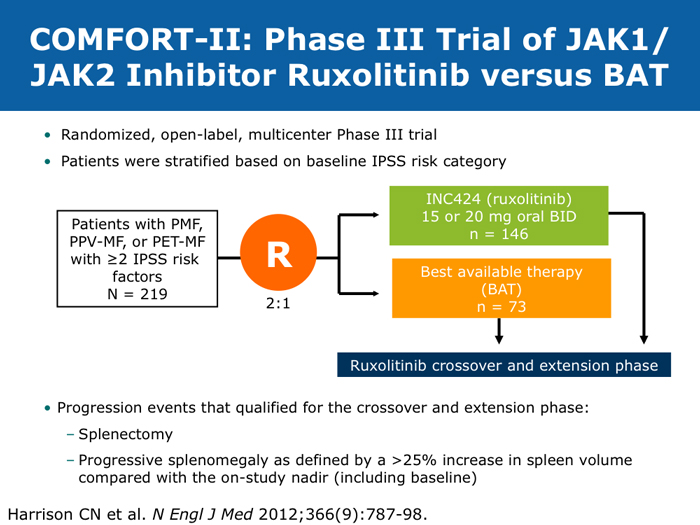 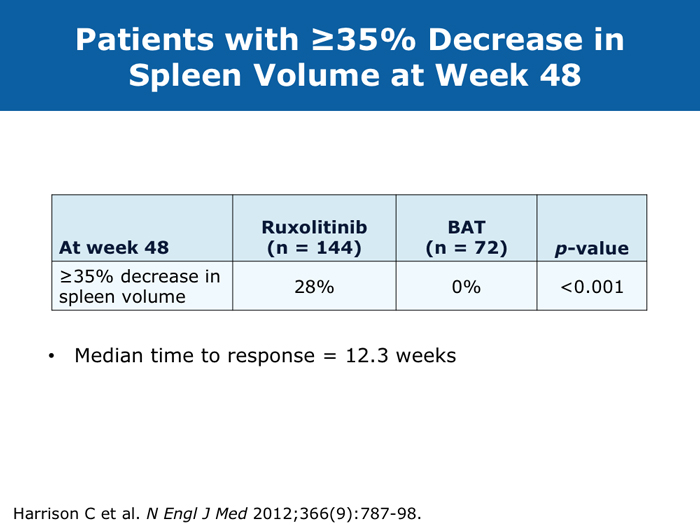  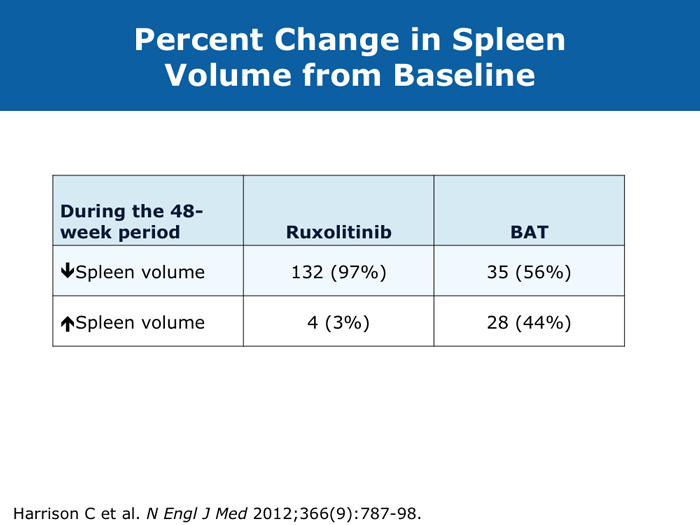 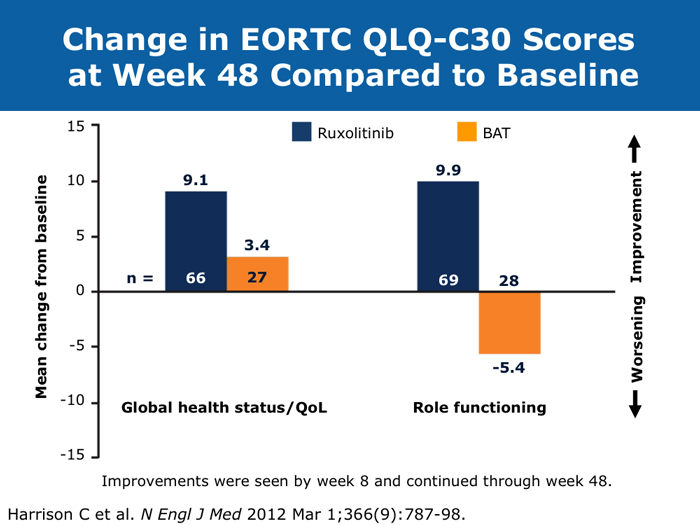  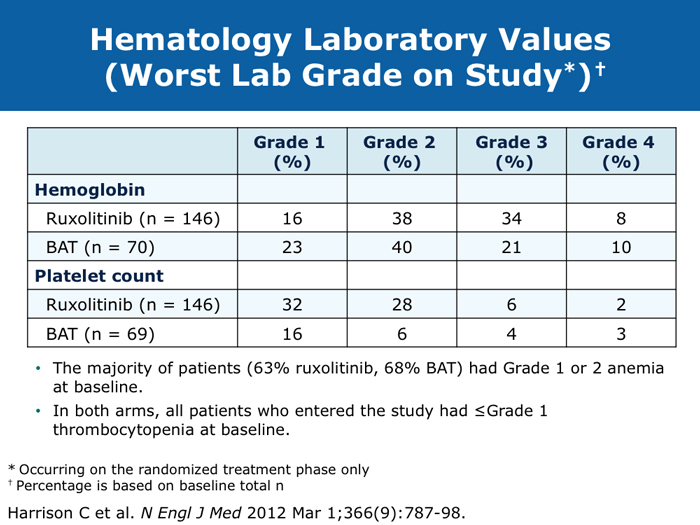 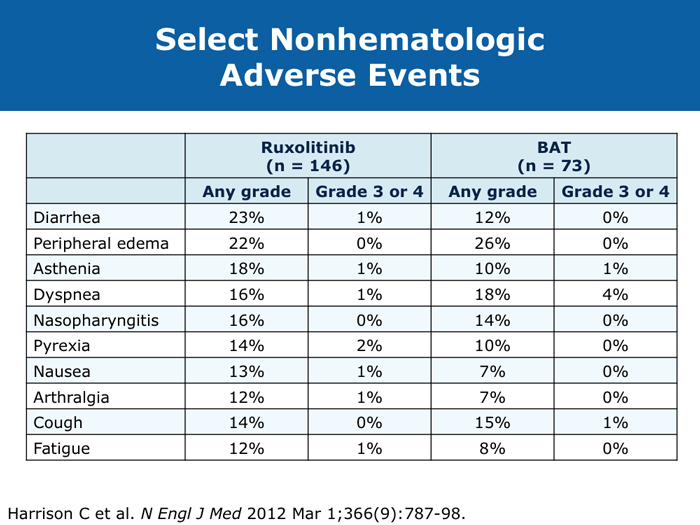 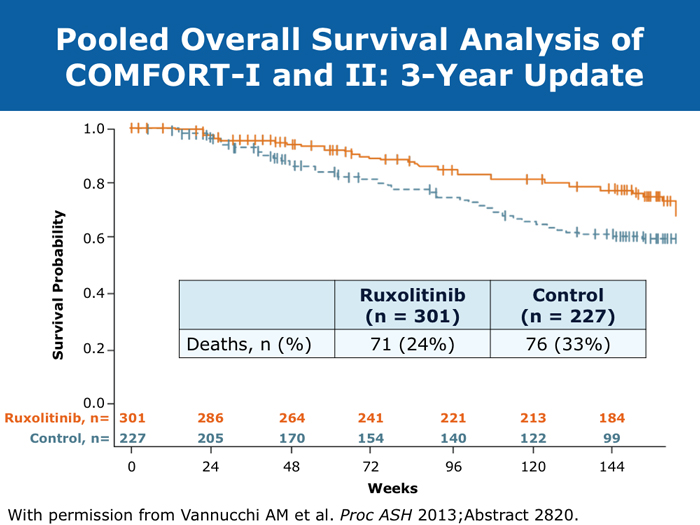  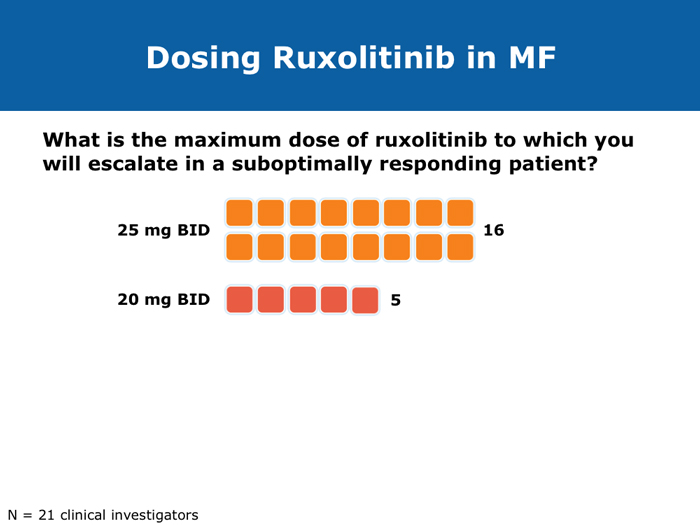  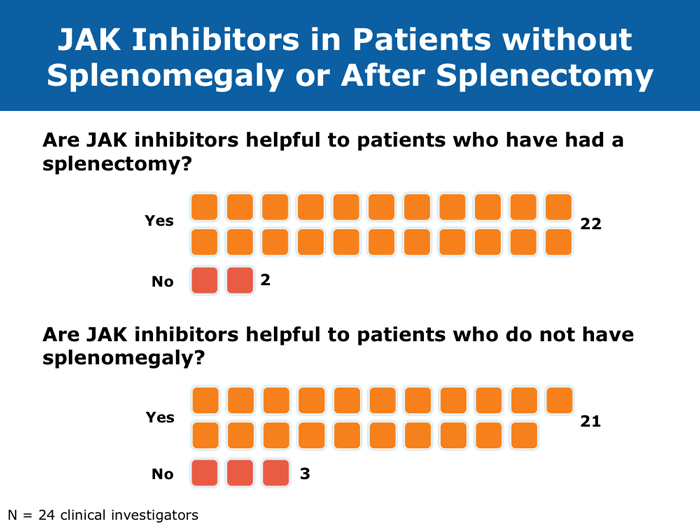 Benjamini O et al. Therapeutic effects of ruxolitinib in patients with myelofibrosis without clinically significant splenomegaly. Blood 2012;120(13):2768-9. Abstract Cervantes F et al; COMFORT-II Investigators. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 2013;122(25):4047-53. Abstract Harrison C et al. Results from a 3.5-year update of COMFORT-II, a phase 3 study comparing ruxolitinib (RUX) with best available therapy (BAT) for the treatment of myelofibrosis. Proc EHA 2014;Abstract P403. Harrison C et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 2012;366(9):787-98. Abstract Tabarroki A et al. Modified dose escalation of ruxolitinib: A feasible therapeutic approach in the management of myelofibrosis. Proc ASH 2013;Abstract 1586. Tefferi A. Challenges facing JAK inhibitor therapy for myeloproliferative neoplasms. N Engl J Med 2012;366(9):844-6. Abstract Vannucchi A et al. A pooled overall survival analysis of the COMFORT studies: 2 randomized phase 3 trials of ruxolitinib for the treatment of myelofibrosis. Proc ASH 2013;Abstract 2820. Verstovsek S et al. Long-term outcomes of ruxolitinib therapy in patients with myelofibrosis: 3-year update from COMFORT-I. Proc ASH 2013;Abstract 396. Verstovsek S et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366(9):799-807. Abstract Anemia can be a challenge but usually does not preclude successful treatment (9 faculty)
Jason Gotlib, MD, MS DR GOTLIB: If patients develop anemia after starting ruxolitinib, it usually occurs by the end of the first month, not during the first week or two. If their hemoglobin drops from 11 to 9 g/dL, that doesn’t cause concern as long as they are not experiencing more fatigue. If their hemoglobin decreases from 10 to <8 g/dL and they are complaining of tiredness, that requires assessing the balance between the improvement in symptoms that they experience with ruxolitinib, which is often dramatic, and the fatigue. I discuss the possibility of transfusion with patients. If they undergo transfusion and the hemoglobin remains low and they are still tired, then I consider reducing the dose of ruxolitinib. If I started them at 20 mg BID, then I’ll reduce the dose to 15 mg BID. I monitor them, and if the hemoglobin starts to rise, then I consider dose escalation. If the patient continues to need transfusions, then I explore adding another agent, such as epoetin alfa or darbepoetin alfa, to ruxolitinib. Ruben A Mesa, MD DR MESA: In practice anemia is probably the largest issue with ruxolitinib use. Hemoglobin level is the most significant driver of how we initially dose ruxolitinib. The natural cutoffs would be hemoglobin levels of 10 g/dL and lower, hemoglobin levels of 8 to 10 g/dL and transfusion dependence. For patients who are not anemic, the highest dose of ruxolitinib that I would start with is 15 mg BID. For patients with a hemoglobin level lower than 10 g/dL, I would administer 10 mg BID of ruxolitinib. If a patient is transfusion dependent, we typically start at 5 mg BID and increase the dose. I tend to make my decision about the starting dose and “ride it out” for the first 3 months. Jumping around with dose reductions in the first several weeks is probably not ideal. I consider anemia to be a somewhat known, expected and sometimes transient toxicity associated with ruxolitinib. If I’ve made the commitment to try the medication for the patient’s benefit, I don’t adjust the dose in the first few weeks simply because of anemia. Srdan Verstovsek, MD, PhD DR VERSTOVSEK: When you administer ruxolitinib at the recommended dose, approximately one quarter of patients develop significant anemia. Proactive monitoring during the first couple of months, when anemia is known to occur, and avoiding treatment interruptions are important because maintaining the exposure to ruxolitinib is ideal. If you stop treatment, the symptoms come back within 10 days. If you see patients only once a month, a patient might develop significant myelosuppression. At that point the physician may say, “Oh. Now he’s too low. We’ll stop ruxolitinib.” Then all the benefits will be lost. The better way would be to decrease the dose and administer a transfusion. Another transfusion may be needed in 2 weeks, but then the red blood cell count will have rebounded because you lowered the ruxolitinib dose and the benefit is not lost. For example, for a symptomatic patient with a hemoglobin level of about 10 g/dL and a platelet count of 220K/µL who was started on 20 mg BID of ruxolitinib but developed anemia after 2 weeks of therapy, I would dose reduce to 10 mg BID and perform a blood transfusion. A proactive decrease and an acceptance of occasional transfusions while patients are recovering at the lower dose are desirable because of the rebound in the red blood cell count. 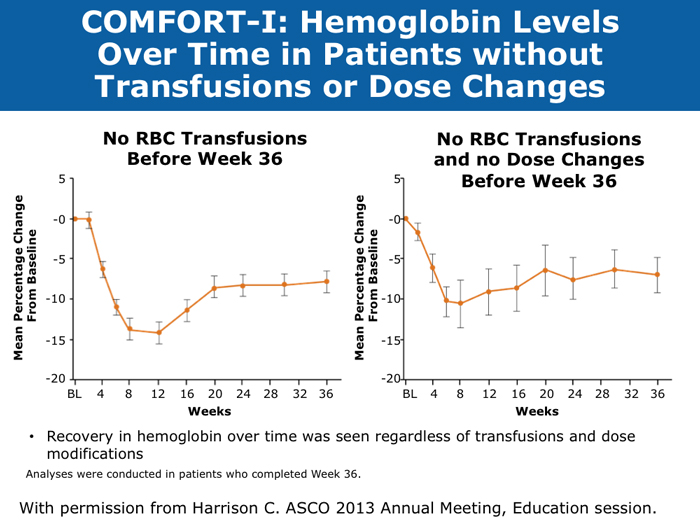    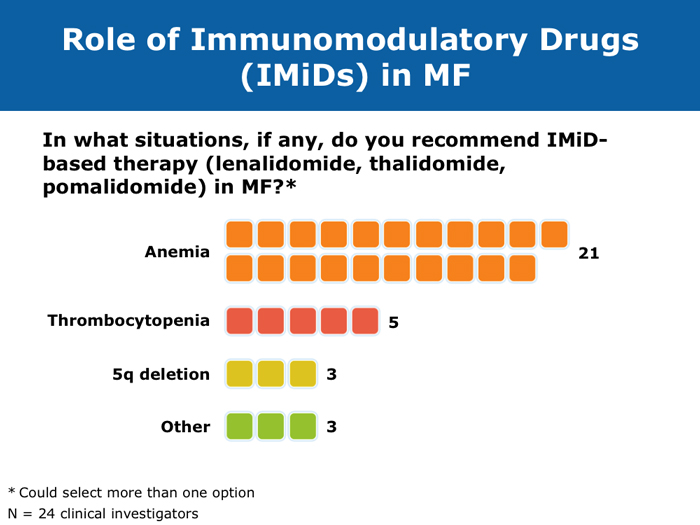 Guglielmelli P, Vannucchi AM. Struggling with myelofibrosis-associated anemia. Leuk Res 2013;37(11):1429-31. Abstract Mascarenhas J et al. An open-label, phase II study of the JAK1 inhibitor INCB039110 in patients with myelofibrosis. Proc ASH 2013;Abstract 663. Mesa R et al. Optimizing dose titration of ruxolitinib: The COMFORT-I experience. Proc ASH 2013;Abstract 4062. Pardanani A et al. Update on the long-term efficacy and safety of momelotinib, a JAK1 and JAK2 inhibitor, for the treatment of myelofibrosis. Proc ASH 2013;Abstract 108. Tabarroki A, Tiu RV. Immunomodulatory agents in myelofibrosis. Expert Opin Investig Drugs 2012;21(8):1141-54. Abstract Verstovsek S et al. Long-term outcomes of ruxolitinib therapy in patients with myelofibrosis: 3-year update from COMFORT-I. Proc ASH 2013;Abstract 396. Select patients with platelet counts as low as 50,000 can successfully receive treatment with a cautious dose-escalation approach (10 faculty)
Moshe Talpaz, MD DR TALPAZ: Both the COMFORT-I and COMFORT-II studies were open to patients with platelet counts over 100,000/µL. At ASH 2012 I presented the results of Study 258, which used a starting dose of 5 mg BID of ruxolitinib and allowed escalation based on platelet counts at monthly intervals for all patients. We included patients with platelet counts of 50 to 100K/µL, which means moderate — 50 to 75K/µL platelets — and mild — 75 to 100K/µL platelets — thrombocytopenia. The take-home message from the study was that the vast majority of patients can receive treatment if they have platelets at the starting point of that range between 50 and 100K/µL, and the average patient ended up receiving 10 mg BID. The responses were slightly lower than the ones observed in the COMFORT-I and COMFORT-II trials but not dramatically lower. About 35% of the patients still experienced a reduction in spleen volume to a level of partial response, and about 40% of the patients experienced a 50% or higher reduction in symptoms. Another benefit was much less prominent anemia. A second interesting finding is that a quarter of the patients with thrombocytopenia may exhibit a splenic sequestration phenomenon, especially if they have early-stage disease. Paradoxically, their platelet counts increased instead of decreasing and they responded beautifully. You need to be aware of this patient group with relatively early disease and those with intermediate-1 or low-risk disease whose low platelet count may be reversed with ruxolitinib. A third observation is that the drop in platelets is highly predictable, usually about 20 to 30K/µL, so you can plan for this. If a patient starts with 50K/µL, then you’ll require treatment interruptions. If a patient starts with platelet counts between 50 and 100K/µL, you usually don’t need to interrupt treatment. It’s predictable. I have administered ruxolitinib to patients with platelet counts lower than 50K/µL. However, the concern is that most of these patients already have damaged bone marrow. They have low cellularity and heavy fibrosis, and the platelet counts continue to drop. So I suggest checking the bone marrow and the cellularity first and being careful not to administer a myelosuppressive treatment to patients with an extremely low bone marrow cellularity of 30% or less. It will not be beneficial. John O Mascarenhas, MDDR MASCARENHAS: It’s well established from the COMFORT-I and COMFORT-II studies that patients with platelet counts higher than 100K/µL can receive ruxolitinib. According to data presented from Study 258, it is also feasible to administer ruxolitinib to patients with platelet counts of 50 to 100K/µL. My recommendation is to start low and titrate upward. I wouldn’t recommend starting ruxolitinib when the platelet count is lower than 50K/µL. With a platelet count of 50 to 100K/µL, I start at 5 mg BID and slowly increase on a monthly basis. At times I titrate up so that the patient receives 5 mg in the morning and 10 mg in the evening. I adopt a stepwise and careful approach. With platelet counts of 100 to 150K/µL, I tend to use 10 mg BID. I follow up with these patients weekly for the first 1 to 2 months to avoid abrupt cessation of the agent. Typically, within the first month of beginning ruxolitinib their platelet counts will give you a sense of whether you will be able to maintain the dose or how quickly you may need to reduce it. 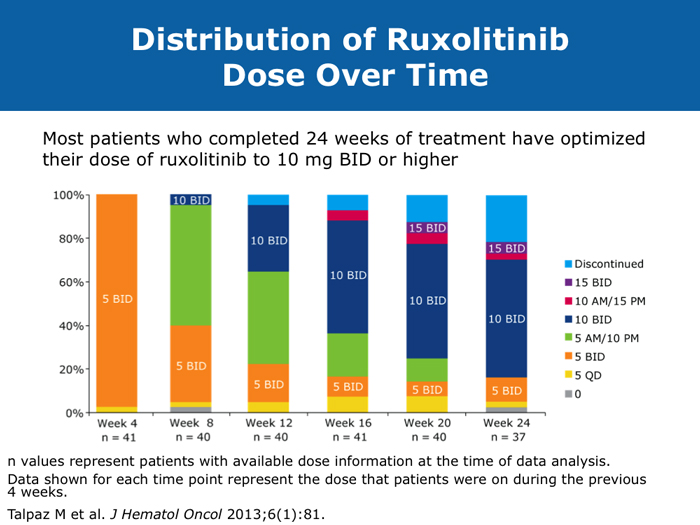 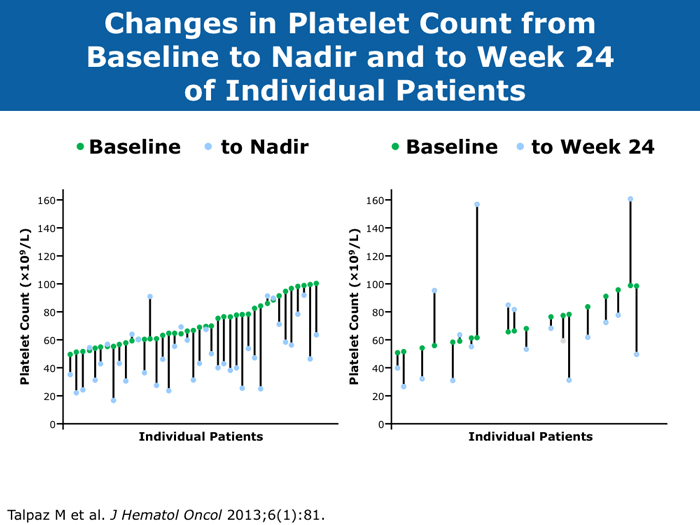  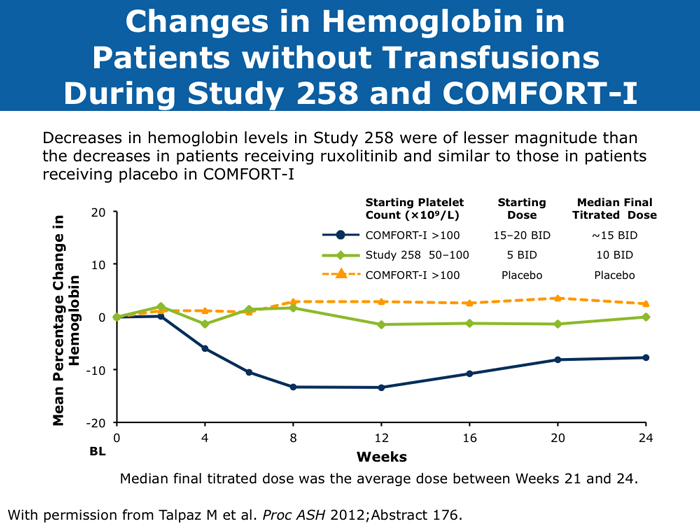 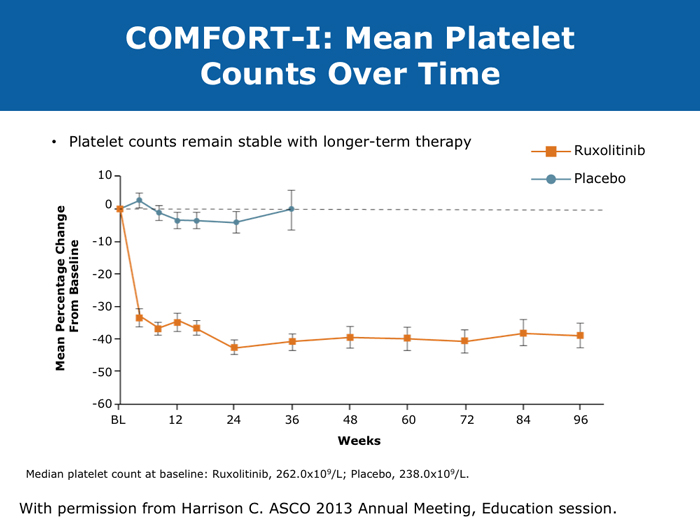 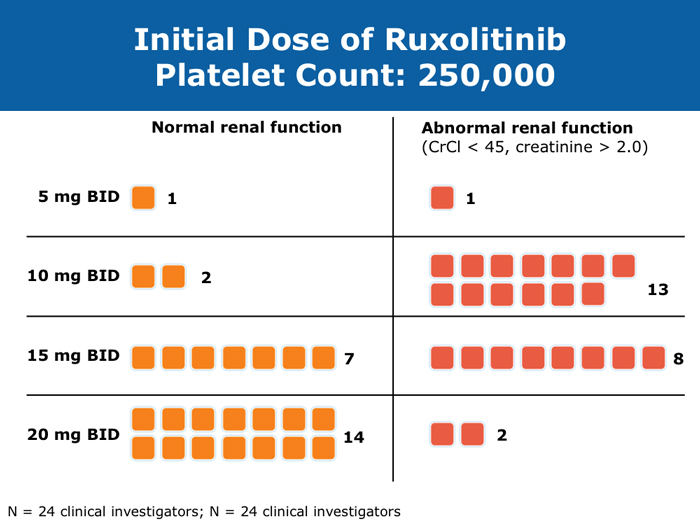 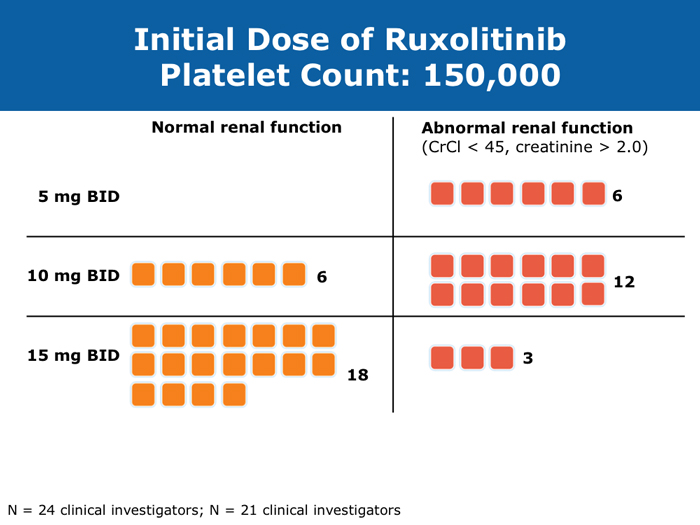 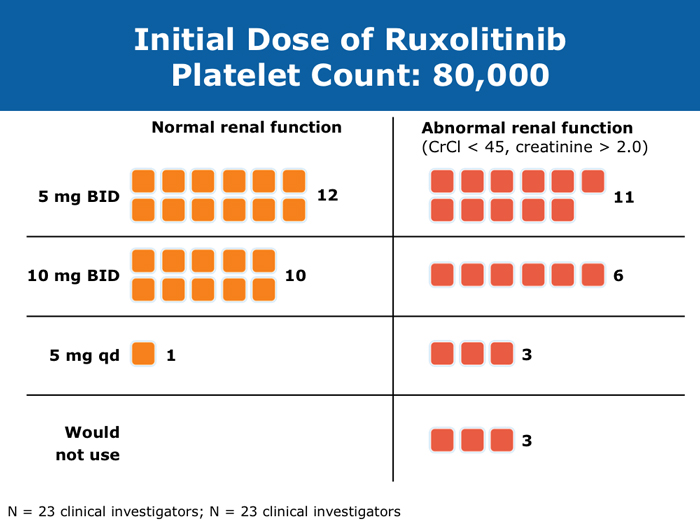  Harrison C et al. Expand: A phase 1b, open-label, dose-finding study of ruxolitinib in patients with myelofibrosis and baseline platelet counts between 50 × 109/L and 99 × 109/L. Proc ASH 2012;Abstract 177. Mesa R et al. Optimizing dose titration of ruxolitinib: The COMFORT-I experience. Proc ASH 2013;Abstract 4062. Talpaz M et al. Interim analysis of safety and efficacy of ruxolitinib in patients with myelofibrosis and low platelet counts. J Hematol Oncol 2013;6(1):81. Abstract Verstovsek S et al. Long-term outcomes of ruxolitinib therapy in patients with myelofibrosis: 3-year update from COMFORT-I. Proc ASH 2013;Abstract 396. Clinical disease progression doesn’t necessarily mandate stoppage of JIs
When stopping therapy, consider tapering More JIs are on the way (9 faculty) Jorge E Cortes, MDDR LOVE: How do you care for patients who have had a very good response to ruxolitinib but then develop splenic regrowth that does not reach pretreatment size? For example, a patient started on ruxolitinib who experiences initial improvement in symptoms and a decrease in spleen size from 18 cm to 2 cm and then presents with an enlarged spleen at 6 cm but still feels well? DR CORTES: That’s an important question, and it’s something that I see with some frequency. It may provide an explanation for the difference between reports that say that 90% of patients eventually discontinue ruxolitinib and those that say that less than 50% eventually discontinue it. My take is that this patient is still experiencing benefit. It’s a lower response compared to the best response that the patient had — the spleen was 2 cm and is now 6 cm — but it’s still an improvement. The patient is feeling well — still eating and has no symptoms — so from my perspective this patient needs to continue therapy. We tend to see a little fluctuation in the spleen size in patients with myelofibrosis who are receiving ruxolitinib. As long as the patient is still feeling good and the spleen is within an acceptable size range, I don’t modify the therapy. To me, these patients are still experiencing a good response, benefiting from the drug, and, in addition, I don’t have a better alternative to offer them. Ruben A Mesa, MDDR LOVE: For a patient in whom you are discontinuing ruxolitinib because of either cytopenias or disease progression, if you are able to taper treatment, do you do that or simply stop? DR MESA: In general, when I discontinue ruxolitinib I taper it. Initially we had concern about “ruxolitinib withdrawal syndrome.” Now that we have significant experience with ruxolitinib in myelofibrosis, we recognize that this is an infrequent issue. If you stop ruxolitinib, the spleen will regrow and patients will feel unwell if you do it abruptly. Patients fare better if ruxolitinib is tapered. Retrospectively, as we’ve observed patients who experienced an aggressive rebound after stopping the drug, we have realized that in the early Phase I studies, if someone became septic for whatever reason — patients who enrolled on these studies were sick — and their JAK inhibitor treatment was stopped, that was a bad combination. However, in general, that’s a rare circumstance. John O Mascarenhas, MDDR MASCARENHAS: Symptoms associated with the discontinuation of ruxolitinib have been an area of controversy. In the COMFORT-I and COMFORT-II studies symptoms returned to baseline within 7 to 10 days of stopping. This was predictable. A single-institution study reported that patients who stopped treatment abruptly developed withdrawal syndrome, which in 1 case was a sepsis-like state. In my experience, symptoms rebound. My practice is to try to taper treatment when I can. If I have to stop abruptly, I almost always use a prednisone taper to blunt the return of symptoms. Regarding cross-resistance with other JAK inhibitors in development, emerging data from several of the ongoing trials indicate that if a patient’s disease doesn’t respond to ruxolitinib, that doesn’t necessarily mean it won’t respond to some of the other JAK inhibitors, probably because there’s promiscuity in their targets. It’s not universal that response or lack of response to one means that no response will be achieved with others. 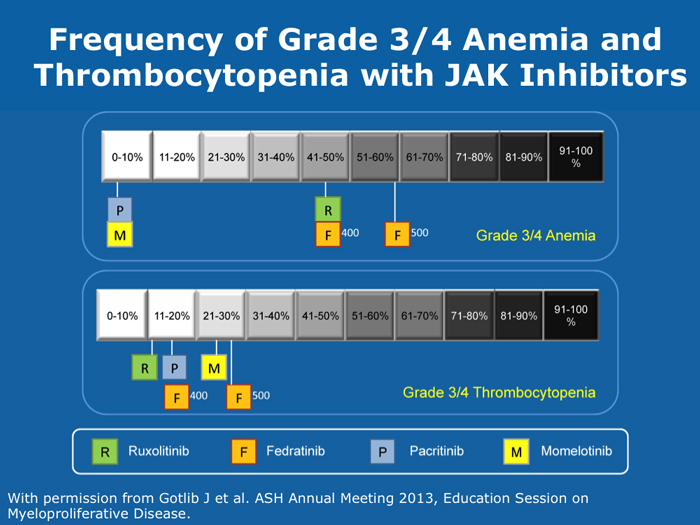 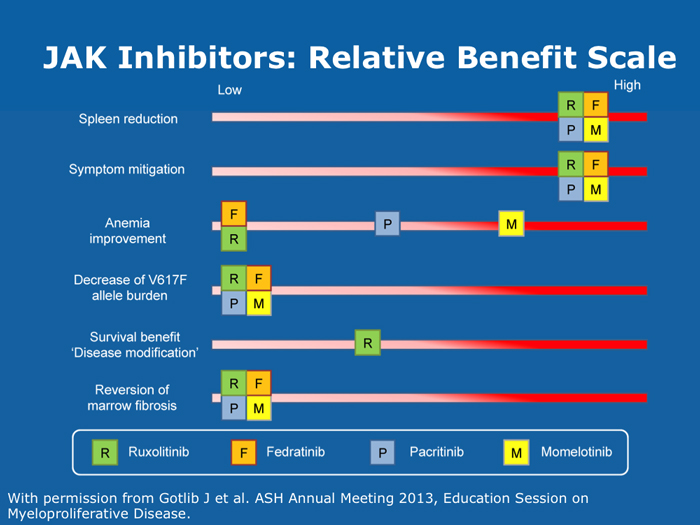  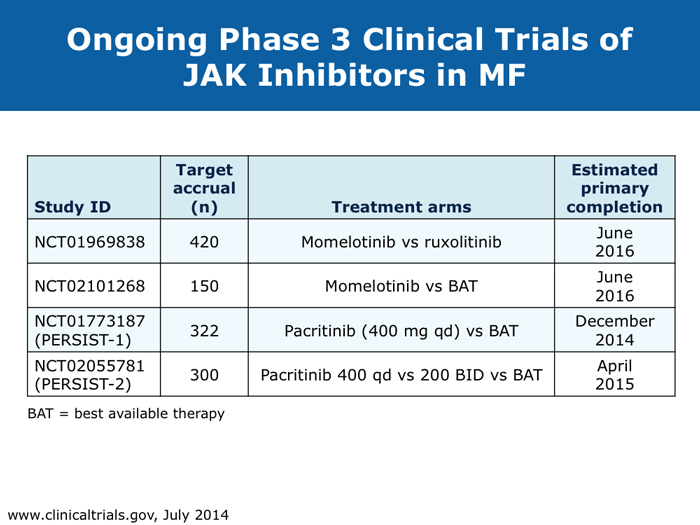 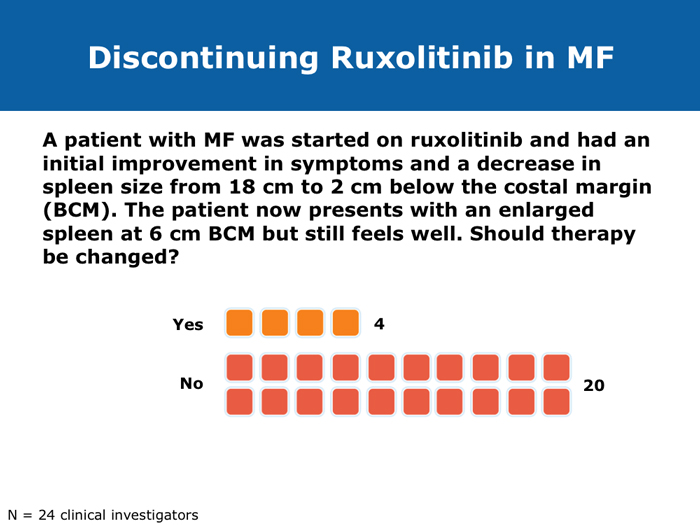 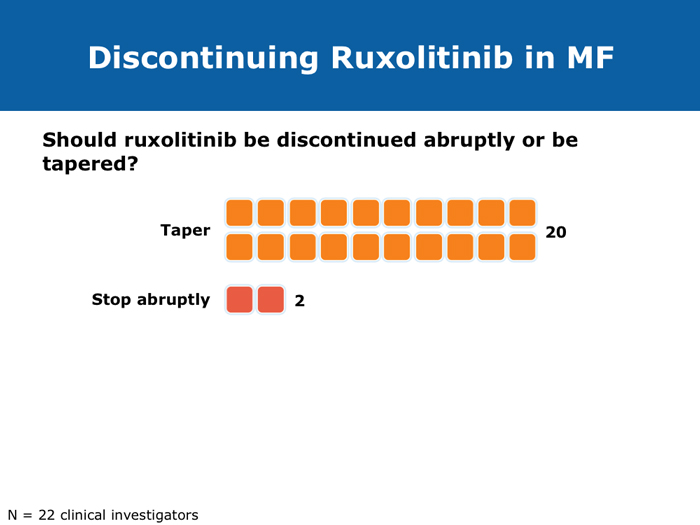  Abdelrahman RA et al. Clonal evolution as determined by sequential bone marrow karyotype analysis during JAK inhibitor therapy for myelofibrosis: Impact on treatment response and overall and leukemia-free survival. Proc ASH 2013;Abstract 2821. Fonseca E et al. Ruxolitinib discontinuation in patients with myelofibrosis: An analysis from clinical practice. Proc ASH 2013;Abstract 2833. Mascarenhas J et al. An open-label, phase II study of the JAK1 inhibitor INCB039110 in patients with myelofibrosis. Proc ASH 2013;Abstract 663. Naito H et al. NS-018, a selective JAK2V617F inhibitor, improves JAK2V617F-induced murine myelofibrosis without decreasing the erythrocyte or platelet count. Proc ASH 2013;Abstract 3847. Pardanani A et al. BMS-911543, a selective JAK2 inhibitor: A multicenter phase 1/2a study in myelofibrosis. Proc ASH 2013;Abstract 664. Pardanani A et al. Retrospective comparison of survival and leukemic transformation in myelofibrosis patients treated with ruxolitinib versus momelotinib versus fedratinib versus pomalidomide. Proc ASH 2013;Abstract 4049. Pardanani A et al. Update on the long-term efficacy and safety of momelotinib, a JAK1 and JAK2 inhibitor, for the treatment of myelofibrosis. Proc ASH 2013;Abstract 108. Sanofi discontinues clinical development of investigational JAK2 agent fedratinib (SAR302503) [press release]. Paris, France, November 18, 2013. Available at: http://en.sanofi.com/Images/34935_20131118_JAK-2-FEDRATINIB_en.pdf. Tefferi A, Pardanani A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin Proc 2011;86(12):1188-91. Abstract Verstovsek S et al. Pacritinib, a dual JAK2/FLT3 inhibitor: An integrated efficacy and safety analysis of phase II trial data in patients with primary and secondary myelofibrosis (mf) and platelet counts ≤100,000/µl. Proc ASH 2013;Abstract 395. Verstovsek S et al. Phase I study of LY2784544, a JAK2 selective inhibitor, in patients with myelofibrosis (MF), polycythemia vera (PV), and essential thrombocythemia (ET). Proc ASH 2013;Abstract 665. |
David P Steensma, MD
DR STEENSMA: When I have given presentations that have included discussion of JAK2 mutations or ruxolitinib or other JAK2 inhibitors, one of the most common questions has been, does the patient need to have the JAK2 mutation to benefit from the therapy? So now when I discuss this I specifically emphasize that you don’t need the mutation — that the pathway is activated even in people without mutations — and I try to hit that head on because I believe that is one of the most common misconceptions about this class of drugs.
DR LOVE: So would you agree or disagree with the statement that regardless of JAK mutation status, the JAK pathway is generally activated in patients with myelofibrosis and that is related to why these patients benefit from ruxolitinib or other JAK inhibitors?
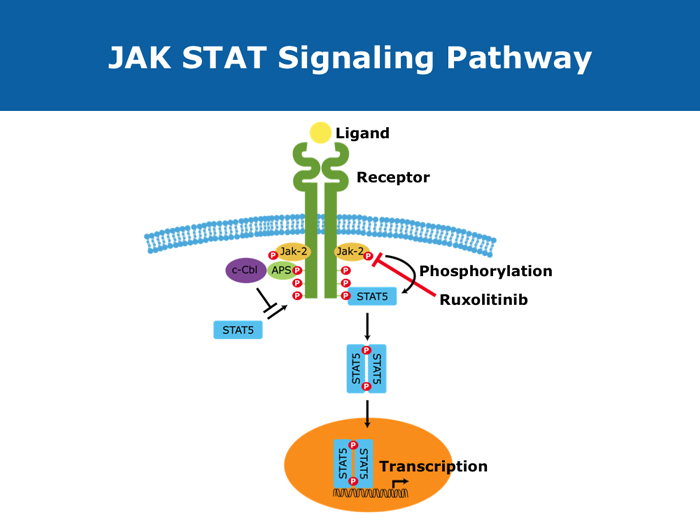
DR STEENSMA: I believe so, yes. I believe that all patients have excessive signaling through that pathway or through its downstream mediators such as AKT, PI3 kinase and the STAT proteins. Downregulating JAK2 signaling and decreasing the excessive cytokine release is beneficial to the patient. That may be the main benefit from these drugs — decreased cytokine production rather than any inhibition of the clone like the inhibition that occurs with imatinib or another tyrosine kinase inhibitor. I believe that is a big part of why patients respond to this drug.
Ruben A Mesa, MD
DR LOVE: How do you conceptualize how the JAK2 inhibitors work, and why do patients without the JAK2 mutations benefit?
DR MESA: This is a key question. My understanding of the process at this point is that all of the JAK2 inhibitors that have been tested are inhibitors of native JAK2. They inhibit JAK2 across the board and not only the mutant form. Now this is beneficial in that JAK2 is overly active in all patients with myeloproliferative neoplasms, whether they have the JAK2 V617F mutation or other mutations. For example, some individuals have mutations in the MPL gene that signals through JAK2, some individuals have mutations in the exon 12 region of the JAK2 gene and in others we might not have identified what mutations they have but those mutations still seem to feed through JAK2.
JAK2 seems to be a central clearinghouse in both groups of patients, and that’s what the drugs inhibit and why we see a benefit. It remains as speculation whether a drug that targeted only the native JAK2 would be more active or less active. But the fact that patients respond with or without JAK2 mutations fits well with what we know about native JAK2 and the fact that it is overly active in both groups of patients.
DR LOVE: When you consider the downstream consequences of inhibiting JAK2, do you specifically think of cytokines?
DR MESA: The overactivation of JAK2 does more than increase cytokine levels. The overactivation of JAK2 is why patients experience myeloproliferation. Whether the cytokines are separate or linked to it, I don’t believe we have enough information yet to know.
DR LOVE: Do you generally believe that ruxolitinib is slowing down the progression of the disease or that it is addressing the secondary consequences and clinical symptomatology?
DR MESA: I believe it has an effect on slowing down the underlying cancer. Myelofibrosis is a glacial disease in certain ways. We are able to measure those features that are the most apparent and immediate, such as splenic symptoms, and then with time perhaps we can measure a delay in the progression of the disease. We’ve noted improvements in survival, and it seems that some patients derive long-term benefit. I believe we have a tendency to underestimate the benefits of ruxolitinib simply because the immediate effects are so obvious.